| TCGAデータベースでRNA | 您所在的位置:网站首页 › GDC Data Portal Homepage › TCGAデータベースでRNA |
TCGAデータベースでRNA
|
RNA-Seqとは
RNA-Seq(リボ核酸シークエンシング)は、リボ核酸(RNA)の配列を高速に決定し、発現量を定量化するための強力な技術です。この技術は、生物の遺伝的情報の流れを理解するための重要なツールとして広く用いられています。 またRNA-Seqでは次世代シークエンシング(NGS)技術を利用して、細胞内の全RNA(全トランスクリプトーム)の配列を決定します。このプロセスは、特定の時間点での遺伝子の発現パターンや、異なる条件下での発現変動を詳細に解析することを可能にします。 RNA-Seqデータ解析のプロセスは、複雑で多段階にわたる作業であり、以下のステップから構成されています。 データの取得 シーケンスされたデータは通常FASTQフォーマットのファイルとして取得でき、アダプタ配列のトリミングや低クオリティのリードの除去などを行います。 マッピング シークエンシングリードを参照ゲノムまたはトランスクリプトームにマッピングします。このプロセスでは、リードの配列が参照配列のどの部分に一致するかを特定します。 マッピング結果をもとにしたリードカウント マッピングの結果を基に、各遺伝子にマッピングされたリードの数をカウントします。これにより、各遺伝子の発現量を定量化します。 発現変動遺伝子の検出 異なるサンプル間で発現量に顕著な変化がある遺伝子を同定します。これは疾患研究や生物学的機能の解析において重要です。今回はマッピング結果をもとにしたリードカウントのデータをダウンロードして、DEG解析、生存予測の解析を行います。 環境 google colacoratory python3.10.12 GDC Data PortalGDC Data PortalとはNIH(アメリカ国立衛生研究所)によるがんゲノムプロジェクトで、多様ながん種について、様々なデータを集約、公開しているものです。 様々な大規模ながんゲノムプロジェクトからのデータを含んでおり、ゲノム配列データだけでなく、転写物レベルデータ、エピゲノムデータ、プロテオミクスデータなど、多様なタイプのがん関連データが含まれています。また、ゲノムデータに加え患者の臨床情報や病理学的データも提供されています。これにより、ゲノム変異と臨床結果との関連についても簡単にアクセスでき、研究者にとって非常に有用なサイトとなっています。 GDC Data Portal
まずはGDC Data Portalを開きRepositoryを開きましょう。
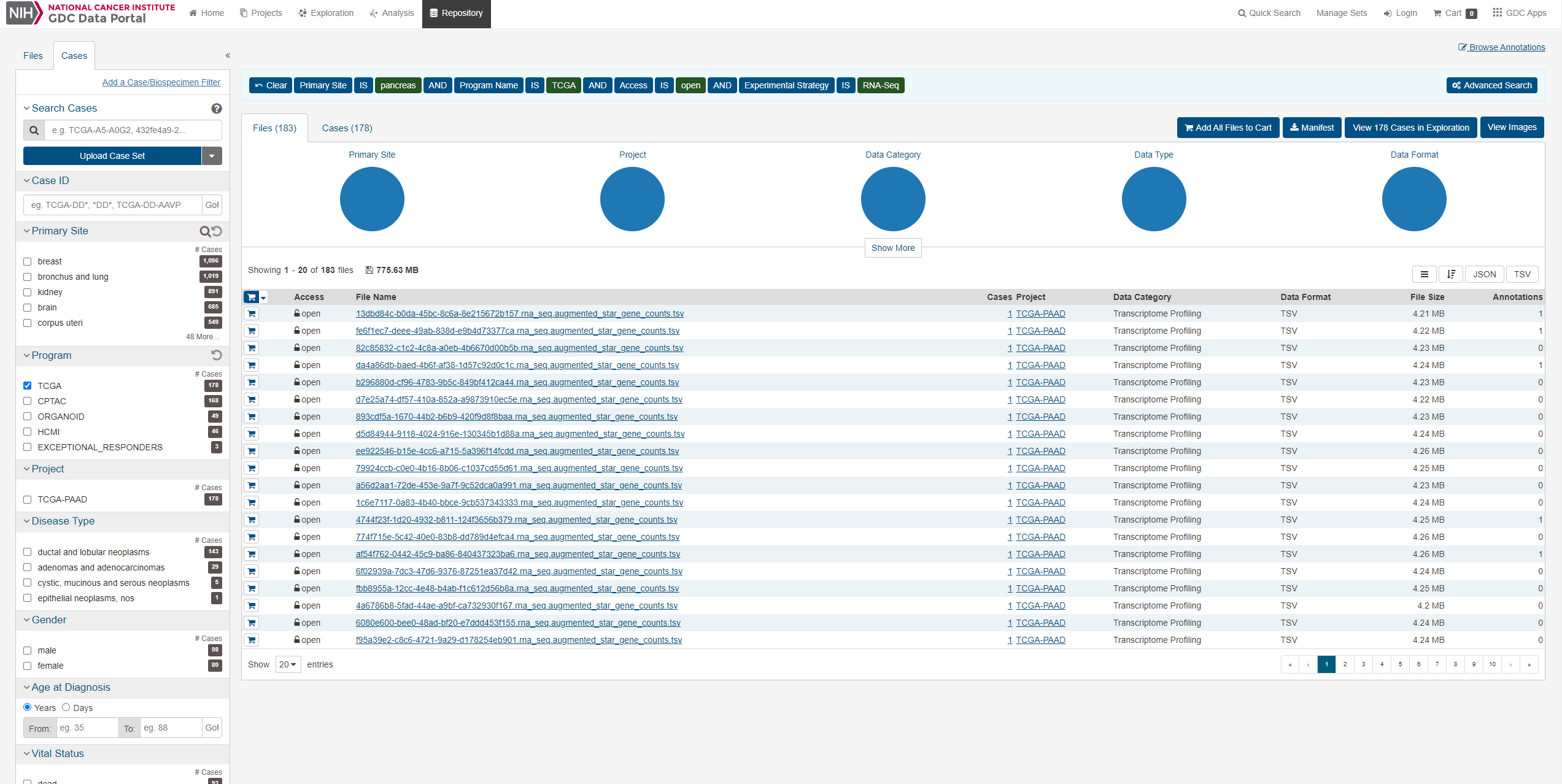
すると以下の画面が出てきます。
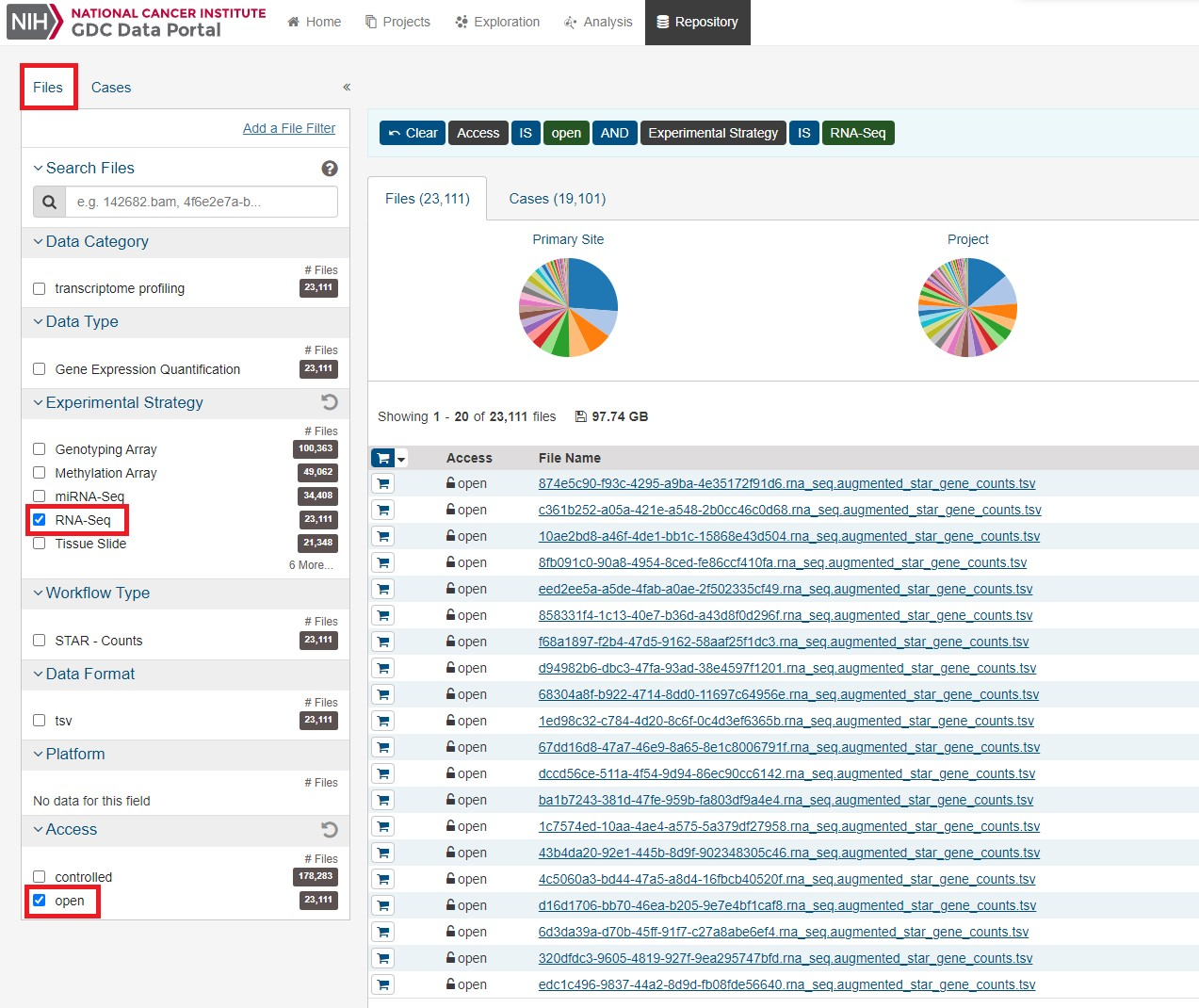
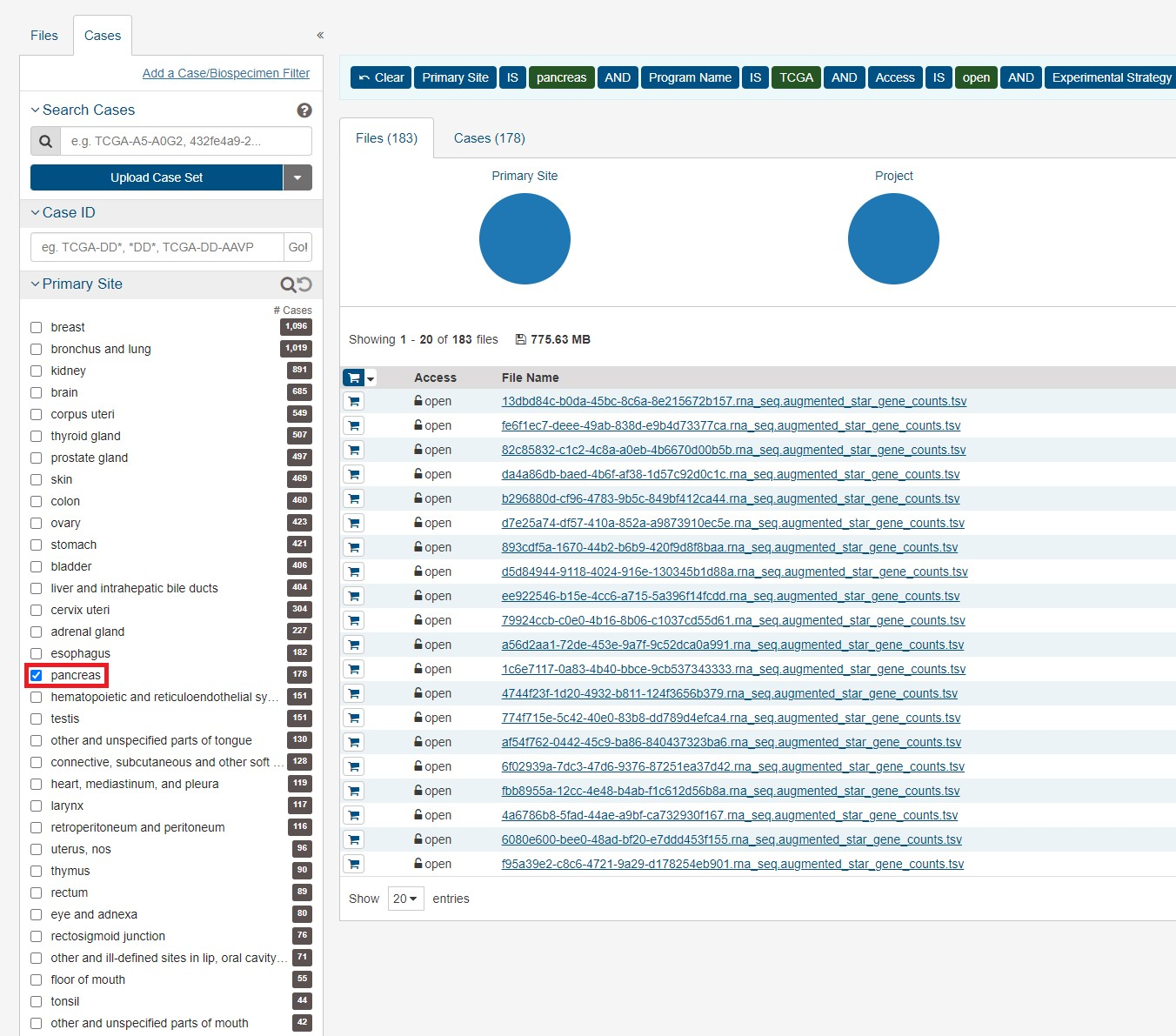
今回は膵臓がんのデータをダウンロードしていきます。データ数のことかく まずはFileタブのRNA-Seqとopenを選択します。
次にCasesタブを開き、今回は膵臓がんなのでpancreasを選択します。
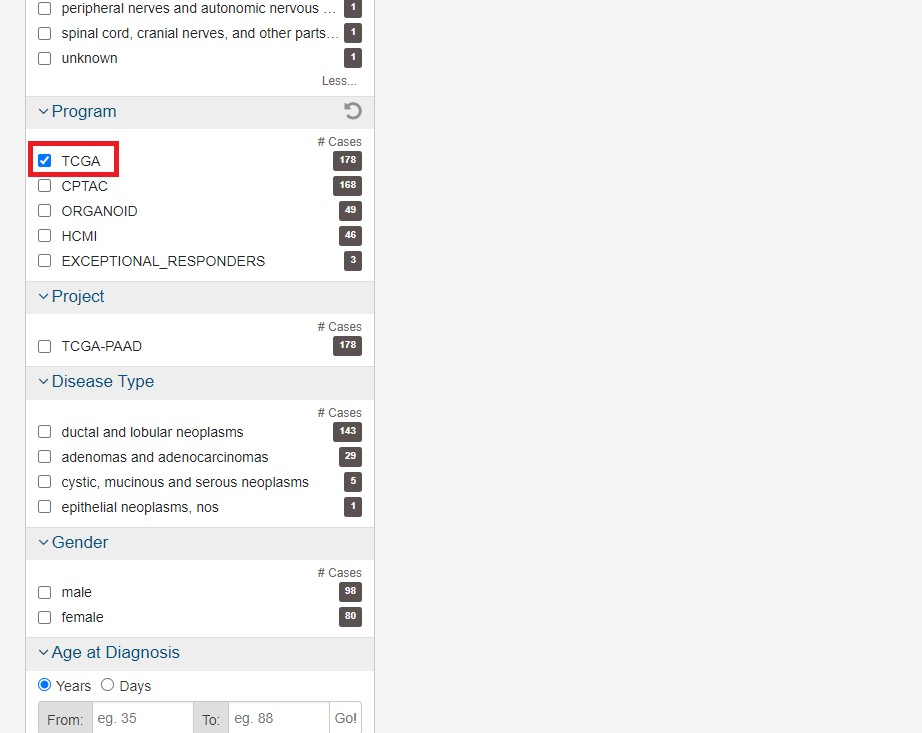
またプログラムからTCGAを選択します ここで様々な条件を指定することができますが、(人種、年齢、性別等)今回はこれらの条件を指定せず進めます。
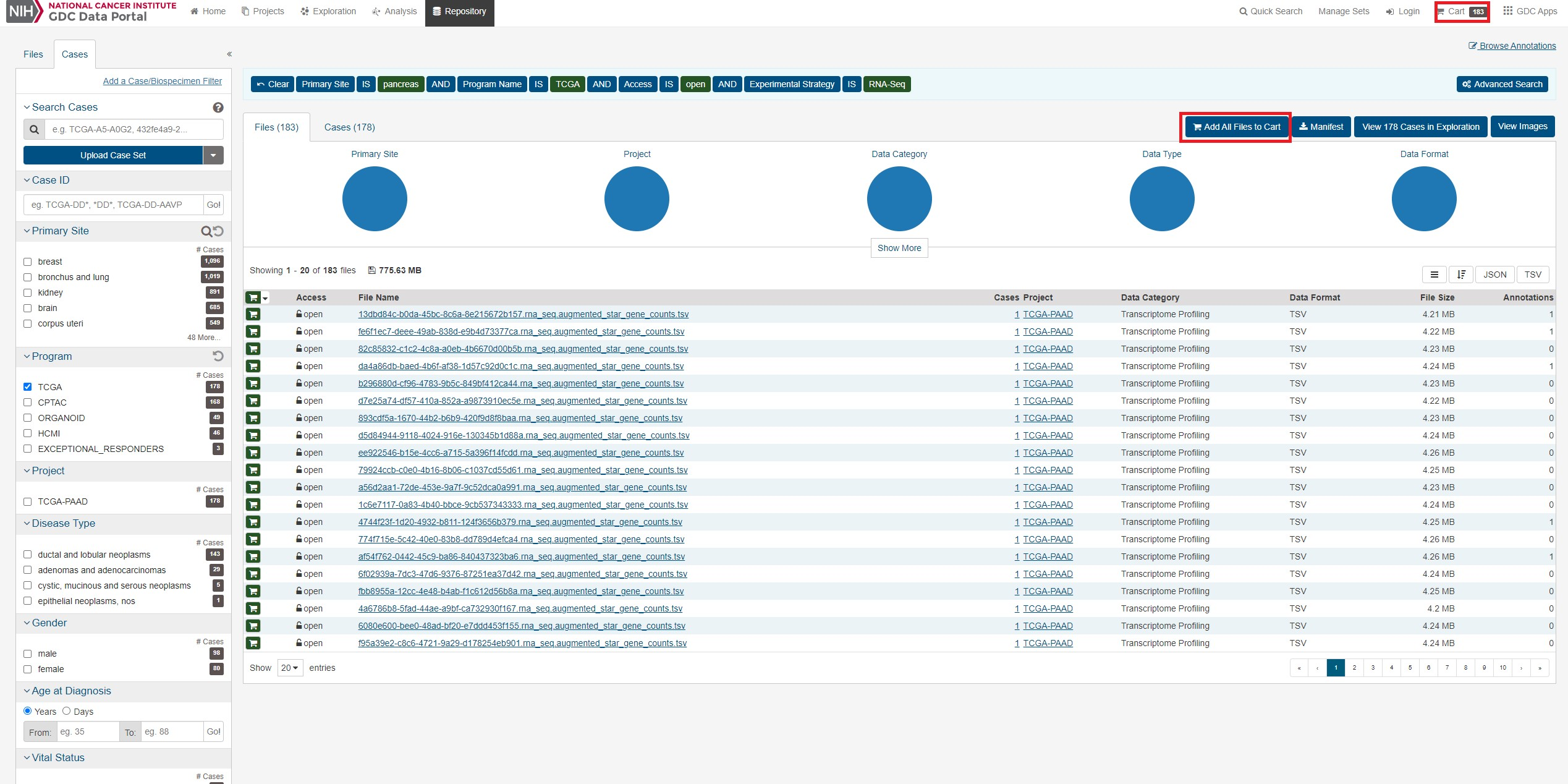
こうして条件を指定できたらすべてのファイルをカートにいれ、カートに行きます
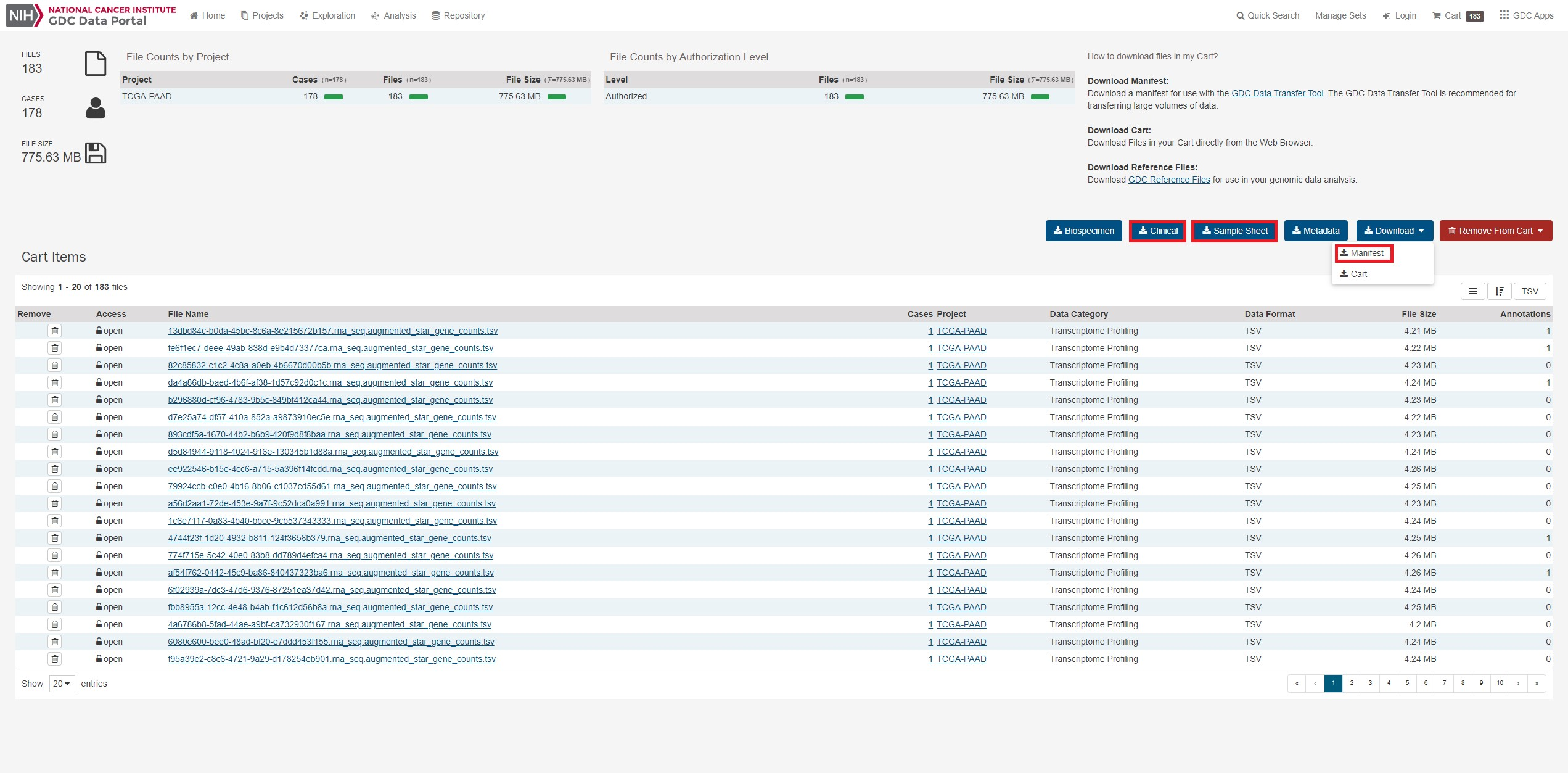
Downloadからmanifestをクリックし、ファイルリストをダウンロードします。 また後ほど必要になるのでsample sheet,clinicalもダウンロードします。
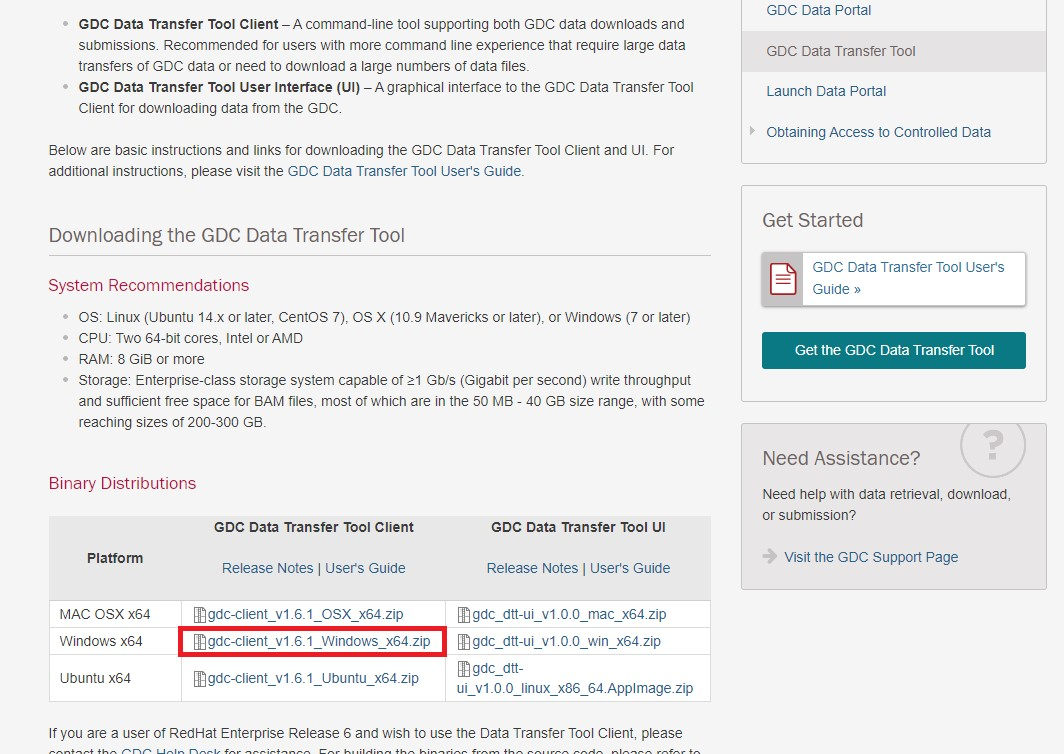
詳しくはマニュアルを参照してください。 GDC Data Transfer Pageに飛び、左のClientから自身の環境にあったものを選びダウンロードしましょう。
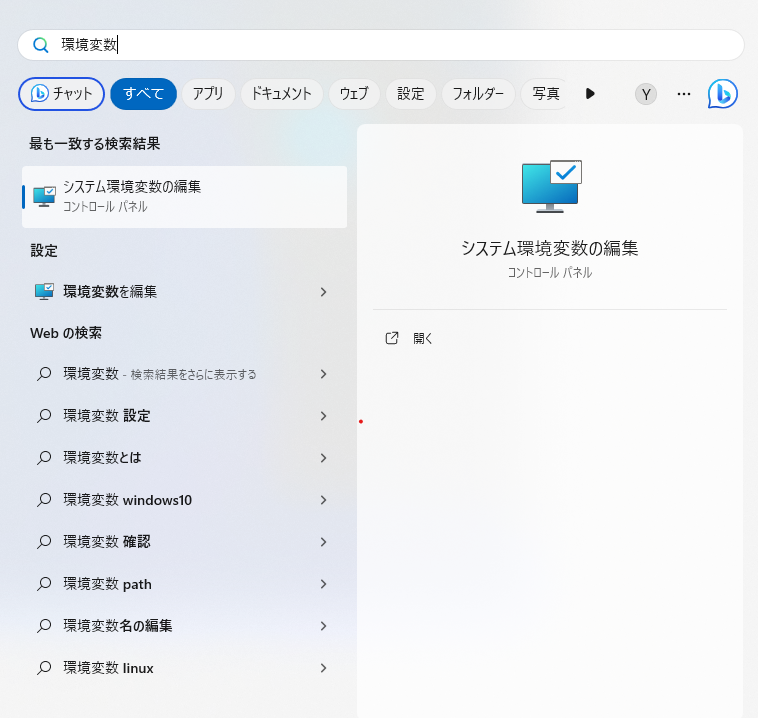
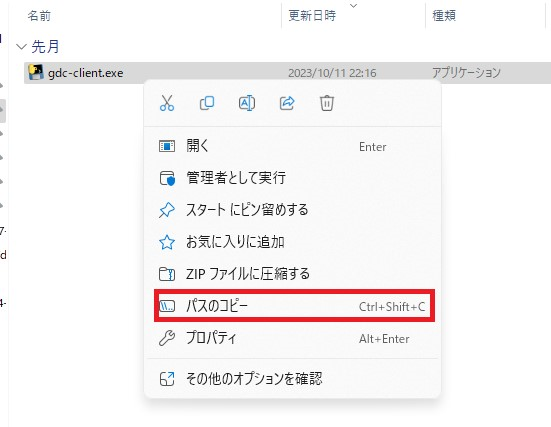
ダウンロードが終わったらパスを通す必要があります。これはホームディレクトリにあるgdc-clientを認識可能にするためのものです。 実行ファイル(exeファイル)を配置した場所の環境変数の設定を行っていきましょう。 まずWindowsキーを押して環境変数と入力し、システム環境変数の編集を開きます。
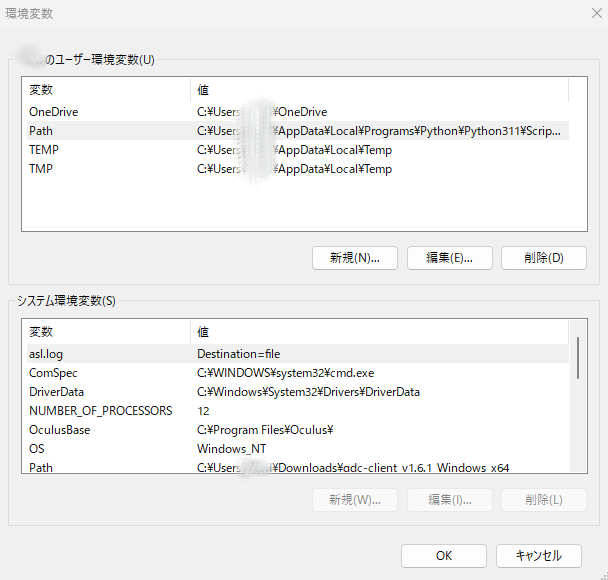
次にユーザー環境変数のPathの編集を押します。
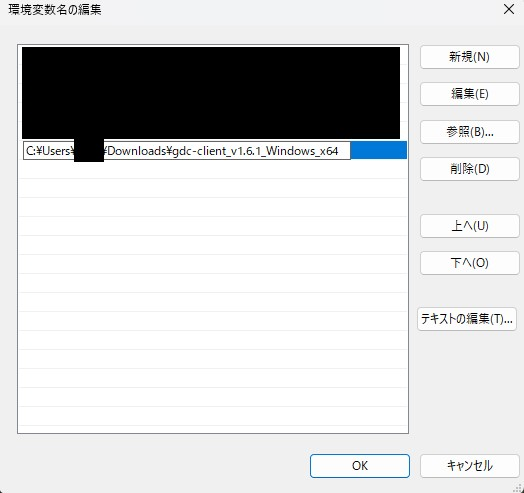
新規を押し、実行ファイルが置かれているフォルダのパスをコピーし貼り付けます。
今回のパスは例えば次の場所からコピーすることができます。
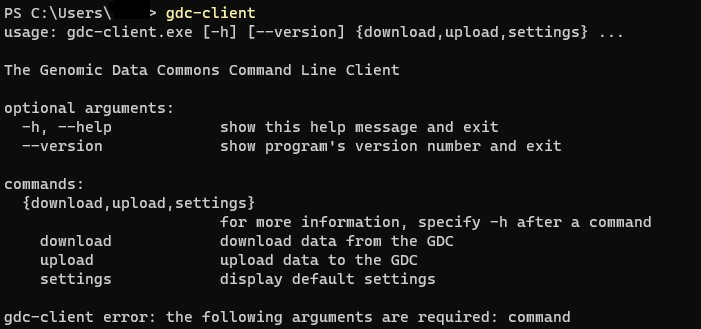
これでパスを通すことができました。 パスが通っているか確認します。 Windowsキーを押してcmdと打ち込みます。そこにgdc-clientと打ち、エラーが出なければ大丈夫です。
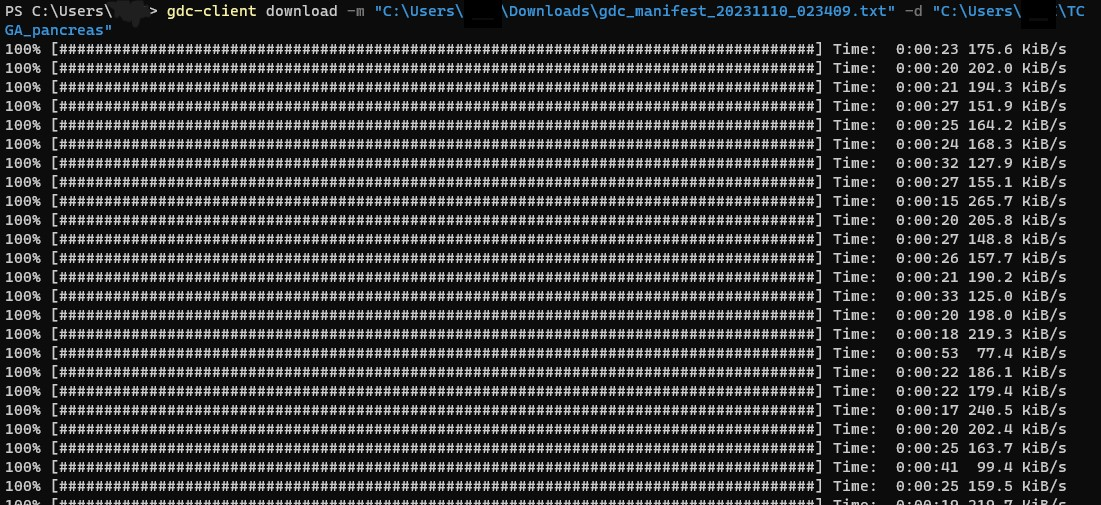
ここからようやくデータのダウンロードに入っていきます。コマンドプロンプト(cmd)を開き、 mkdir TCGA_pancreasと適当なフォルダを作ります。ここにダウンロードしたデータを入れていきます。名前はTCGA_pancreasとしました。 -mオプションをつけてManifestファイルのパスを指定し、-dオプションで保存先ディレクトリ(今作成したTCGA_pancreasフォルダ)を指定します。 gdc-client download -m "Manifestファイルのパス" -d "保存先ディレクトリのパス"するとダウンロードが始まります。今回は200個弱あり、それなりに時間がかかります。
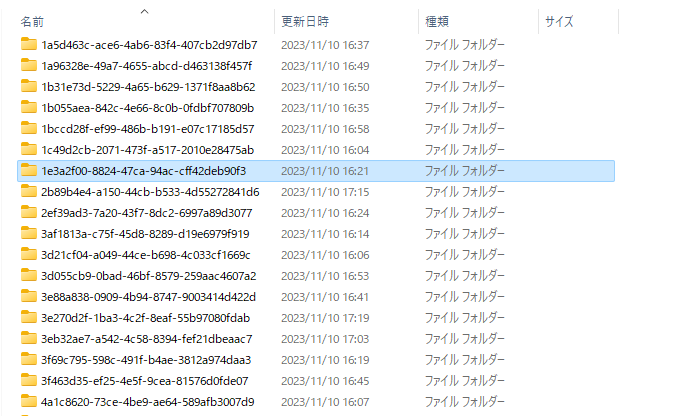
ダウンロードが終わるとTCGA_pancreasフォルダにこのようにデータがダウンロードされました。
中身を見てみましょう。下にtsvファイルがあり、ここに欲しいデータがあります。
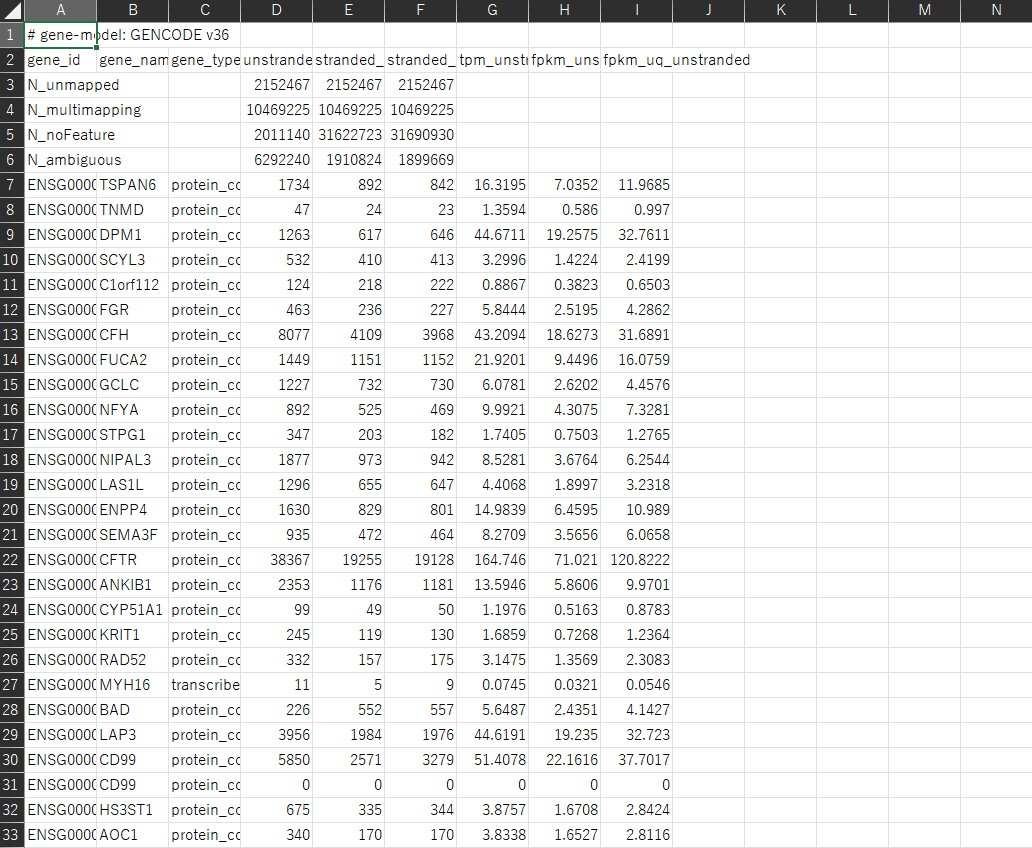
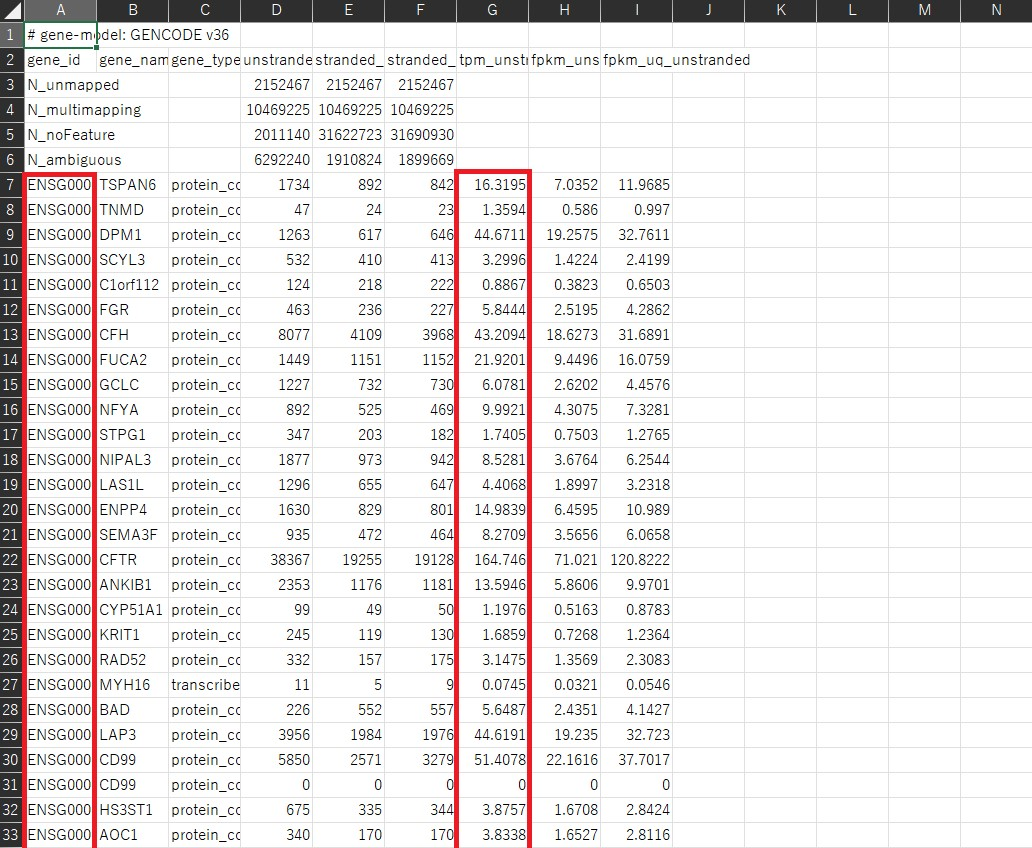
これはExcelで開いたものですが、データはこのようになっています。 今回は発現量カウントの補正方法としてTPMを用います。発現量カウントの補正方法については以下の記事が参考になると思います。 遺伝子発現量カウントの補正方法(RPM, RPKM, TPM)【Python】
以上の様に遺伝子発現量のデータをダウンロードすることができましたが、このままでは解析を行いにくいのでpython のデータ解析ライブラリであるpandasライブラリに対応するために、一つののDataFrameにデータをまとめていきます。 目標は以下の様なDataFrameを作ることです。行に遺伝子、列に検体が並びます。今回は60660行183列の表が出来上がります。
今回はgoogle colaboratoryで作業を行います。 まず作成したTCGA_pancreasフォルダをGoogle Drive上にアップロードします。新規を押して、フォルダをアップロードでTCGA_pancreasフォルダをアップロードします。 また、今回作業用にTCGA_pancreas_workingフォルダも作り、これもアップロードします。
ここまで来たらここ(google colacoratory)にアクセスしてください。 詳細な作業についてはgoogle colaboratoryで説明するとしてここでは大まかな方針を説明します。 まず各ファイルについて下図の様にgene_idとTPM値のみをとりだします。こうして2列のファイルを作ります。
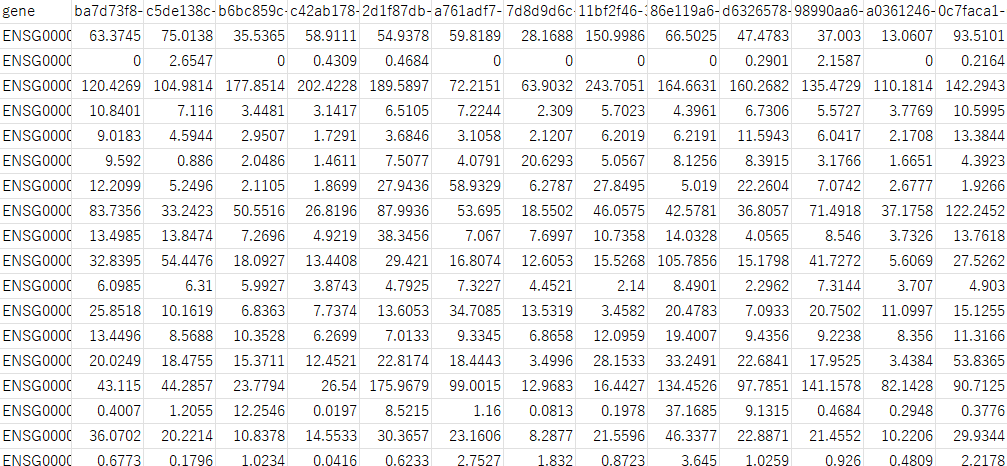
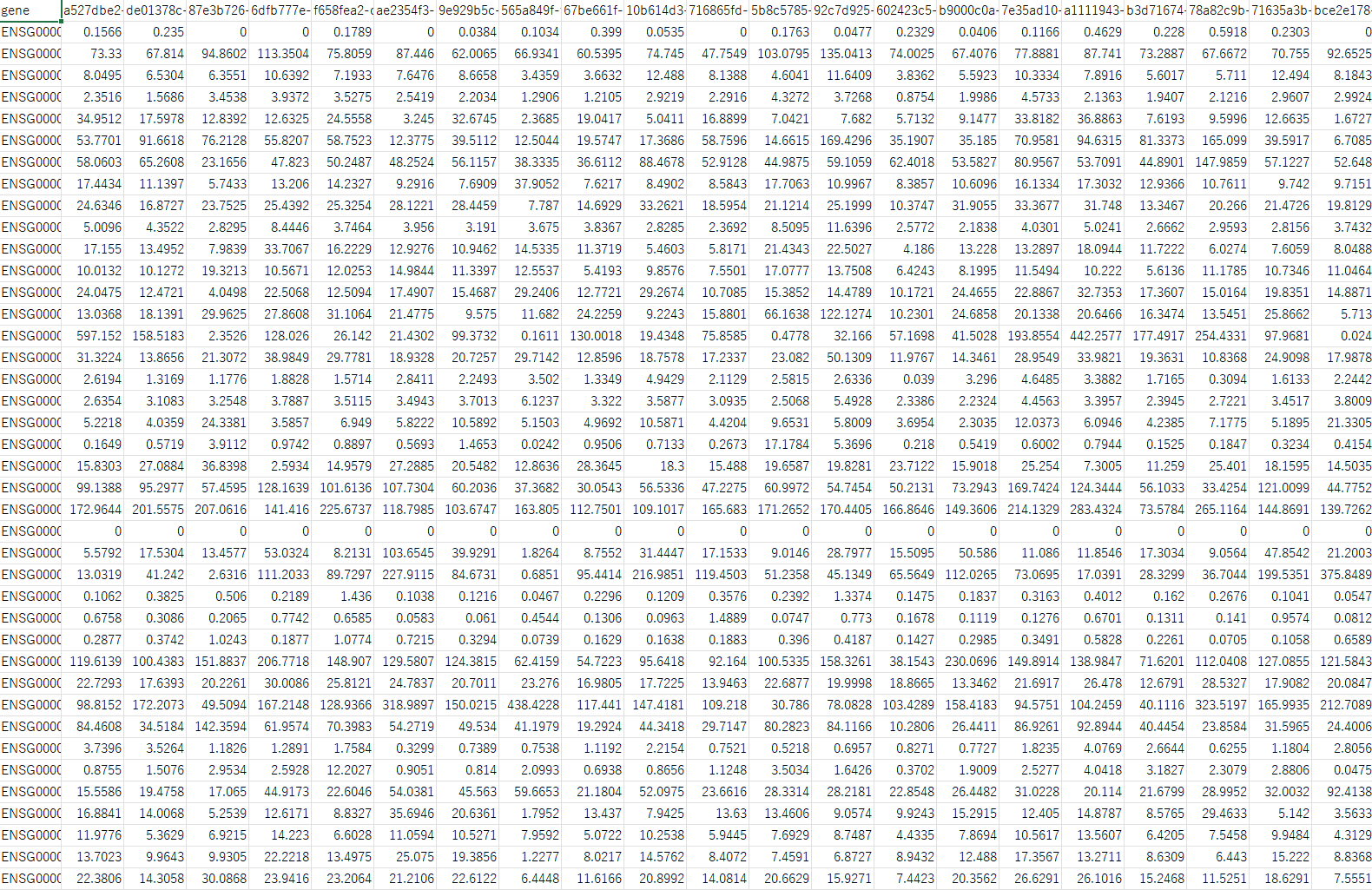
次に検体1の2列目の右側(3列目)に検体2の2列目(TPM値)を追加します。これを検体の数だけ繰り返してDataFrameを作成します。 こうして作成したDataFrameは以下の様になります。
発現変動遺伝子(DEGs, Differentially Expressed Genes)とは、異なる条件やグループ間において発現量が大きく上昇または減少した遺伝子のことを言います。ここではそうした発現変動遺伝子を探していきます。 DEG解析にはpyDESeq2を用います。 pyDESeq2は、PythonでDESeq2を使用してRNAシーケンスデータのDEG (Differential Gene Expression) 分析を行うためのライパーです。DESeq2は、RNAシーケンスデータのための統計的手法で、生物学的レプリケート間の遺伝子発現の変動を考慮しながら、条件間での発現の差異を特定するものです。 今回pyDESeq2において必要となるデータはcountsとmetadataの二つのファイルとなります。 countsとmetadataはそれぞれ以下の様になっています。
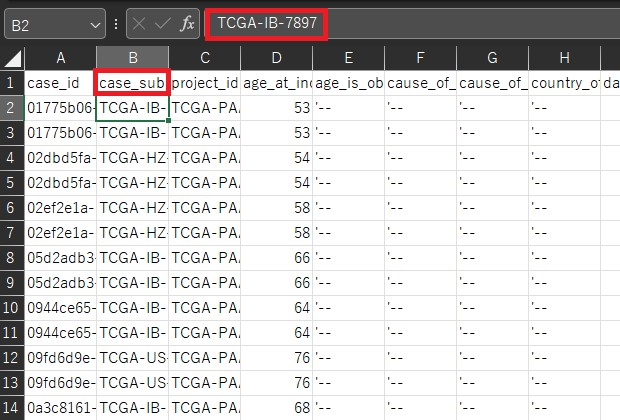
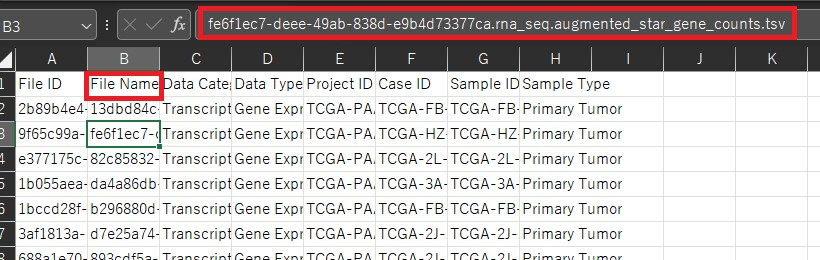
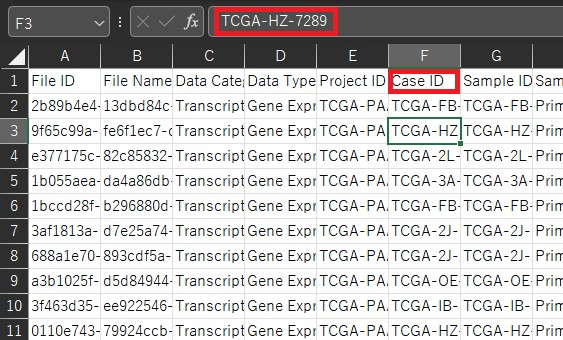
countsはここまでで作ってきたDataFrameを転置したものになります。 metadataは各サンプルの状態が入力されているものです。この例では状態が二つ入力されています。(conditionとgroupの二つ)今回はdead or aliveかで単因子解析を行うのでconditionのみの列となります。 ここで用いるのはGDC Data Portalから発現データをダウンロードした時に一緒にダウンロードしたsample sheetとclinicalです。clinicalはフォルダになっていて中にファイルが何個か入っていますが今回使うのはclinical.tsvです。 このclinicalファイルには様々な状態が記載されていますが、今回は単一因子分析としてDead or Aliveの二つの状態のみに注目したいと思います。 つまり1列目にサンプル名2列目にvital_statusとなるようにファイル操作を行うとclinicalファイルからmetadataを作ることができます。しかしこのサンプル名が先ほど作ったDataFrameとclinicalで違うので、sample sheetを用いてこの二つをつなぐ必要があります。 具体的に見てみましょう。先ほど作成したDataFrameではsample名としてファイル名を用いています。
一方でclinicalではファイル名は存在しておらず、サンプル名としてcase_submitter_idが用いられています。
そこでこのファイル名とcase_submitter_idをつなげるためにsample sheetを用います。sample sheetではファイル名はFile Nameの列に、case_submitter_idはSample IDの列にあります。(clinicalのcase_idとは違います。)
具体的な作業はここ(google colacoratory)で行っています。 countsとmetadataが出来上がったらあとは実際にDEG解析を行っていきます。先に述べたpyDSeq2を用います。 解析の結果を見ていきましょう。下図は結果の一部となります。
発現変動遺伝子を特定するためにpadjが0.01よりも小さいものを取り出します。
機械がいつ故障するか。患者がいつ死亡するか。このようにある事象がいつ起こるのかを推定する手法として生存分析があります。今回はDEG解析で特定した発現変動遺伝子を用いて生存予測を行います。 必要なファイルは学習用データ、評価用データとなります。 サンプル数(患者数)が183名のため、学習用データに100名を用い、評価用データとして83名を用います。 学習用データは目的変数と説明変数の2つにわけることができます。 目的変数はvital_status 説明変数は各遺伝子 となります。つまり、各遺伝子がvital_statusにどの様に関与しているかを学習し、予測します。今回はロジスティック回帰を用いてモデリングを行います。 例のごとく具体的な作業はここ(google colacoratory)で行っています。 モデリングをした後評価用データにあてはめてみます。5つだけ結果を表示します。 pred = model.predict_proba(X_test)[:, 1] print(pred[:5])結果は[2.21084671e-04 9.79174058e-01 2.19425586e-18 1.06323643e-43 9.99999998e-01]となりました。 このようにほぼ1か0のどちらかになってしまうのは、説明変数の数が多すぎることが原因と思われます。つまり100個の変数を3294個の変数で説明するというところに無理があるということです。これは過学習とよばれ、今回もこれに相当すると思われます。 予測結果をAUCで評価しました。AUCとはROC曲線の下の面積を指し、主に分類モデルの性能を評価するために使用されます。AUCは、モデルがクラスをどの程度正確に分類できるかを示す指標です。この数値の目安は以下の様になっています。 0.9~1.0:非常に精度が高い 0.8~0.9:精度が高い 0.7~0.8:多少精度が高い 0.6~0.7:あまり良くない精度 0.5~0.6:ランダムと変わらない精度 from sklearn.metrics import roc_auc_score # AUCを計算 auc = roc_auc_score(true, pred) print(f"AUC: {auc}")今回の結果は0.504となり作成したモデルは予測に全く使えないものとなってしまいました。これの原因は先に述べた過学習にあると考えています。 まとめTCGAから遺伝子発現量を取得し、DEG解析を行いすい臓がんを引き起こす遺伝子を3000個程度特定し、それをもとに生存予測を行いました。 しかしながら生存予測が過学習によりうまくいきませんでした。これは特定したがん遺伝子が3000個と多かったことが原因なのでp値をもっと小さい値で原因遺伝子を選定するとよいと思われます。 今回記事を書くにあたりプログラミングがほぼ初心者だったので全般的に苦労しました。 特に苦労したのはファイル操作(Pandasの扱い)でした。なれないPandasの操作に四苦八苦しました。しかしChatGPTの助けなども得ながら進めることができました。 ご指摘、アドバイス等ございましたら気軽にコメントお願いします。 (この記事は研究室インターンで取り組みました:https://kojima-r.github.io/kojima/) |

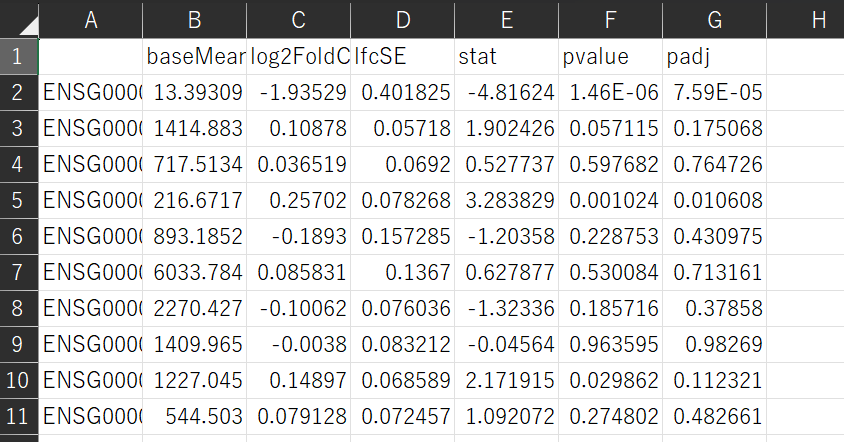
 この様に、3294個の遺伝子を発現変動遺伝子として特定することができました。
この様に、3294個の遺伝子を発現変動遺伝子として特定することができました。【本文地址】